
Caros Colegas e Restantes Leitores este foi um livro feito pelo 24º Curso de Higiene Oral, do qual eu pertenço orgulhosamente. Nele estão decritas resumidamente algumas patologias (deficiências e seus efeitos na cavidade Oral. Espero que seja útil.
1. Agenésia do corpo caloso
|
Descrição Sucinta da Patologia
|
 Má formação congénita do SNC, com aumento significativo dos cornos occipitais. Pode ser uma agenésia total (ausência total do corpo caloso), parcial (ausência parcial do cc), hipoplasia do corpo caloso (corpo caloso de tamanho pequeno) ou disgenésia do corpo caloso (alguns locais com má formações ou sub-desenvolvidos).
Má formação congénita do SNC, com aumento significativo dos cornos occipitais. Pode ser uma agenésia total (ausência total do corpo caloso), parcial (ausência parcial do cc), hipoplasia do corpo caloso (corpo caloso de tamanho pequeno) ou disgenésia do corpo caloso (alguns locais com má formações ou sub-desenvolvidos).Causas: Erros inatos do metabolismo; exposição fetal a toxinas; doenças vasculares e infecciosas; uso abusivo de álcool e cocaína durante a gestação e presença de outros síndromes genéticos.
|
Características Gerais (se houver)
|
Na maioria dos casos é assintomática.
Os sintomassão o síndrome de desconexão cerebral; cefaleias; hemiparesia; hipotonia; malformações oculares; osteoartrites; deficit mental variável e falta de coordenação entre os dois hemisférios.
Características: Atraso variável na aquisição de acções como andar, falar e ler; coordenação motora pobre; sensibilidade atípica a estímulos tácteis e alta tolerância à dor.
Está habitualmente associada a: Anomalias faciais; atraso mental; paralisia cerebral; epilepsia, hidrocefalia; anomalias gastrointestinais e cardiovasculares; hipertelorismo; holoprosencefalia; polimicrogiria; microcefalia e lisencefalia.
|
Características Orais (se houver)
|
Apenas dos medicamentos, normalmente devido a epilepsia associada.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Neurologistas; endocrinologistas; geneticistas; oftalmologistas; terapeutas da fala; terapeutas ocupacionais; fisioterapeutas e assistentes sociais.
2. Meningite Bacteriana
|
Descrição Sucinta da Patologia
|
 Meningite é um processo inflamatório que acomete as meninges (ou mais especificamente as leptomeninges) que são membranas protectoras que revestem o cérebro e a medula espinhal. Agentes patogénicos entram na corrente sanguínea, quando estes se dirigem para o cérebro vão proliferar com grande facilidade, levando a inflamação, edema cerebral que consequentemente conduzem ao aumento da pressão craniana e a sequelas.
Meningite é um processo inflamatório que acomete as meninges (ou mais especificamente as leptomeninges) que são membranas protectoras que revestem o cérebro e a medula espinhal. Agentes patogénicos entram na corrente sanguínea, quando estes se dirigem para o cérebro vão proliferar com grande facilidade, levando a inflamação, edema cerebral que consequentemente conduzem ao aumento da pressão craniana e a sequelas.Causada por: bactérias – neisseria meningitidis (meningococo), streptococus pneumoniae (pneumococo) haemophilus influenza (hemófilo); por vírus ou fungos.
|
Características Gerais (se houver)
|
Os sintomassão variáveis dependendo da idade do indivíduo.
Em recém-nascidos e lactentes: febre, possibilidade de mãos ou pés frios, náuseas, vómitos, choro frequente, irritabilidade, rigidez da nuca e coluna arqueada, expressão parada, dificuldade em despertar e letargia, manchas na pele, abaulamento da fontanela, movimentos involuntários e moleza no corpo.
Em crianças e adultos: rigidez do pescoço, cefaleias, febre, vómitos, fotofobia, sonolência, confusão, dores no corpo e convulsões.
|
Características Orais (se houver)
|
Problemas/Características orais estão associadas as possíveis sequelas e não à patologia em si.
Possíveis sequelas: paralisia cerebral, surdez, cegueira, hidrocefalia, etc.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
As meningites bacterianas são tratadas com antibióticos e corticoesteroides. No caso das meningites virais (mais comuns e menos graves) muitas vezes nem é necessária medicação.
3. Trissomia 18/Síndrome de Edwards
|
Descrição Sucinta da Patologia
|
 A trissomia 18 é um distúrbio genético que consiste na presença de um cromossoma 18 extra no genoma do portador deste distúrbio. Este material genético extra interfere no desenvolvimento normal. Os portadores desta patologia têm uma esperança média de vida muito baixa (morrem geralmente na infância).
A trissomia 18 é um distúrbio genético que consiste na presença de um cromossoma 18 extra no genoma do portador deste distúrbio. Este material genético extra interfere no desenvolvimento normal. Os portadores desta patologia têm uma esperança média de vida muito baixa (morrem geralmente na infância).Causas: Anomalias genéticas herdadas ou adquiridas.
|
Características Gerais (se houver)
|
Atraso mental, atraso no desenvolvimento, malformações do coração, microcefalia, pescoço curto, occipital pronunciado, hipertonia, unhas hipoplásicas, baixo peso à nascença, pernas cruzadas (posição preferida), orelhas abaixo do nível normal, anomalias renais e genitais externos anormais.
|
Características Orais (se houver)
|
Mandíbula pequena, palato alto e estreito, alterações relacionadas com os medicamentos.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Geneticistas; oftalmologistas; terapeutas da fala; terapeutas ocupacionais; fisioterapeutas; neurologistas e cardiologistas. Cirurgias para correcção de mal formações do coração.
4. Síndrome de Cockayne
|
Descrição Sucinta da Patologia
|
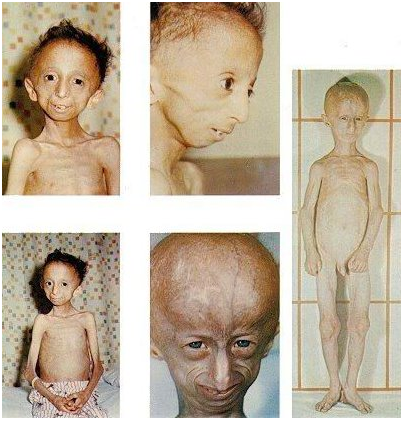 Desordem genética rara de hereditabilidade autossómica recessiva, por mutação no gene ERCC6 ou ERCC8 que codificam proteínas reparadoras de erros genéticos. Quanto mais cedo se manifestar a doença, mais rápida será a sua progressão, maior o número de sintomas e menor a esperança média de vida.
Desordem genética rara de hereditabilidade autossómica recessiva, por mutação no gene ERCC6 ou ERCC8 que codificam proteínas reparadoras de erros genéticos. Quanto mais cedo se manifestar a doença, mais rápida será a sua progressão, maior o número de sintomas e menor a esperança média de vida.|
Características Gerais (se houver)
|
Baixa estatura, microcefalia, envelhecimento precoce, deformações da coluna, problemas de circulação, baixa temperatura, micropénis, testículos recolhidos, olhos encovados, falta de gordura subcutânea facial, nariz estreito e pequeno, fotossensibilidade, atraso no desenvolvimento mental e neurológico moderado a severo, anomalias nutricionais, problemas renais, anomalias no fígado, hipertensão, calcificações intra-cranianas, retinopatias, cataratas, cegueira, perda auditiva, osteoporose, ataxia, etc.
|
Características Orais (se houver)
|
Aumento da cárie dentária, hiperplasia gengival, atrofia dos processos alveolares, agenésia de permanentes, hipoplasia condilar, raízes curtas e cónicas, atraso na esfoliação e erupção dentária e má oclusão.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Fisioterapia, terapia ocupacional, terapia da fala, auditiva, oftalmológica, colocação de sonda gástrica, medicação para problemas cardíacos, hepáticos, renais, etc.
5. Síndrome da Delecção do Cromossoma 4
|
Descrição Sucinta da Patologia
|
 É uma anomalia genética rara, caracterizada por atraso mental e de crescimento e dismorfismo facial típico conhecido como “rosto em capacete grego”, que inclui glabela proeminente, hipertelorismo e nariz em bico, compromete o desenvolvimento e crescimento.
É uma anomalia genética rara, caracterizada por atraso mental e de crescimento e dismorfismo facial típico conhecido como “rosto em capacete grego”, que inclui glabela proeminente, hipertelorismo e nariz em bico, compromete o desenvolvimento e crescimento.A doença afecta a qualidade de vida do paciente e pode ser uma ameaça para ele, podendo levar à morte nos primeiros anos. O peso de nascimento é geralmente inferior a 2Kg apesar de um período gestacional normal ou mesmo prolongado, indicando um atraso de crescimento intra-uterino.
As manifestações são o resultado de uma delecção parcial do braço curto de um cromossoma 4. Muitas crianças com este síndrome morrem durante a infância e as poucas que sobrevivem até a segunda década de vida apresentam deficiências graves e maior risco de infecções e epilepsia.
|
Características Gerais (se houver)
|
Muitos não são capazes de caminhar; maioria não desenvolve uma fala activa, apenas alguns são capazes de fazer frases complexas; microcefalia, hipertelorismo (afastamento excessivo olhos); nariz grande e adunco, pavilhões auriculares grandes, micrognatia (tamanho pequeno da mandíbula inferior); lábio leporino e/ou palato fendido; glabela proeminente, fissuras palpebrais; antimongolóides (olhos caídos), estrabismo; boca de carpa (curvatura da boca voltada para baixo em alguns casos); malformações cardíacas mais frequentemente: defeitos septais, atriais ou ventriculares; anomalias geniturinárias; deformidades ósseas são também comuns e a mais frequente é o pé equino-varu (torto).
|
Características Orais (se houver)
|
Atraso na erupção dentária; agenésia; fusão dentária; lábio leporino ou fenda palatina; microglossia; dentes tendem a ser pequenos e espaçados; dificuldade na sucção e deglutição.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Fisioterapia, terapia ocupacional, terapia da fala. Alguns pacientes necessitam de ajudas físicas, por exemplo, cadeira de rodas, talas, aparelhos auditivos. Pacientes com cardiopatias congénitas, pés tortos, criptorquidia devem ser tratados cirurgicamente. Aqueles com crises recorrentes e necessidade de EEGs – recomendado drogas antiepilépticas.
6. Lisencefalia
|
Descrição Sucinta da Patologia
|
 O termo lisencefalia engloba um grupo de malformações raras, devidas à ausência total ou parcial de circunvoluções no córtex cerebral. Esta deve-se à ocorrência anormal de um dos processos mais críticos no desenvolvimento cerebral – a migração dos neurónios durante a embriogénese. Os indivíduos portadores desta doença morrem, frequentemente, por aspiração de alimentos ou fluidos e por doenças respiratórias. A esperança de vida para estes pacientes está intimamente relacionada com o grau de malformação do cérebro.
O termo lisencefalia engloba um grupo de malformações raras, devidas à ausência total ou parcial de circunvoluções no córtex cerebral. Esta deve-se à ocorrência anormal de um dos processos mais críticos no desenvolvimento cerebral – a migração dos neurónios durante a embriogénese. Os indivíduos portadores desta doença morrem, frequentemente, por aspiração de alimentos ou fluidos e por doenças respiratórias. A esperança de vida para estes pacientes está intimamente relacionada com o grau de malformação do cérebro.|
Características Gerais (se houver)
|
Os indivíduos com lisencefalia apresentam disfagia, anomalias do tónus muscular (hipotonia), convulsões e atrasos psicomotores severos.
|
Características Orais (se houver)
|
Alterações orais relacionadas com a medicação (para controlar as convulsões).
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
A lisencefalia não tem cura. É possível, no entanto, o tratamento sintomático que visa melhorar a qualidade de vida dos indivíduos portadores: as convulsões podem ser controladas através de medicação apropriada; os problemas na deglutição requerem formas adaptativas de alimentação, como a gastrostomia, a fim de prevenir a aspiração de alimentos; os problemas ortopédicos, resultantes da hipotonia, podem também ser atenuados através da fisioterapia articular e respiratória; e o tratamento pode passar, ainda, pela resolução do problema referente ao refluxo gastroesofágico, também frequente nestes indivíduos.
7. SÍndrome do cromossoma x FRÁGIL
|
Descrição Sucinta da Patologia
|
 É uma doença genética ligada ao cromossoma X, este apresenta uma falha na porção subterminal do seu braço longo (Xq27.3), sendo a causa mais frequente da deficiência mental hereditária. Os indivíduos portadores têm uma expansão de 54 a 200 sequências repetidas do codão CGG, que se designa por pré-mutação, os que são clinicamente afectados têm uma mutação completa, que consiste numa expansão que varia de 230 a 2000. Esta últimaestá associada à metilação do gene FMR 1, que bloqueia a transcrição deste e como consequência a proteína FMRP não é produzida. Em condições normais, a FMRP encontra-se em elevados níveis nos neurónios, logo a sua deficiência ou ausência é responsável pelas características clínicas e intelectuais da doença.
É uma doença genética ligada ao cromossoma X, este apresenta uma falha na porção subterminal do seu braço longo (Xq27.3), sendo a causa mais frequente da deficiência mental hereditária. Os indivíduos portadores têm uma expansão de 54 a 200 sequências repetidas do codão CGG, que se designa por pré-mutação, os que são clinicamente afectados têm uma mutação completa, que consiste numa expansão que varia de 230 a 2000. Esta últimaestá associada à metilação do gene FMR 1, que bloqueia a transcrição deste e como consequência a proteína FMRP não é produzida. Em condições normais, a FMRP encontra-se em elevados níveis nos neurónios, logo a sua deficiência ou ausência é responsável pelas características clínicas e intelectuais da doença.|
Características Gerais (se houver)
|
Hiperactividade, movimentos estereotipados das mãos e auto-agressão, défice de concentração e atenção, humor instável, contacto visual escasso e interacção social pobre. Os homens têm um défice cognitivo que varia entre dificuldades de aprendizagem e deficiência mental profunda, já elas apresentam um desenvolvimento cognitivo no limiar da normalidade ou um défice cognitivo ligeiro. Apresentam macrocefalia e hipotonia, a face alongada e estreita, com a mandíbula proeminente, orelhas grandes e “em abano”, macrorquidia, pés chatos, hiperextensibilidade das articulações, escoliose e calosidade das mãos.
Patologias associadas: episódios convulsivos; otites médias agudas recorrentes ou otites serosas; estrabismo, miopia, astigmatismo, hipermetropia e nistagmo; 50% apresentam prolapso da válvula mitral, o que obriga a profilaxia antibiótica.
|
Características Orais (se houver)
|
Apresentam o palato alto e podem ter alterações relacionadas com os fármacos antiepilépticos.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Não tem cura, mas decorrem investigações como a terapia genética, que estuda a possibilidade de inserir no cromossoma X um gene perfeito substituindo o gene FMR1 alterado; pela terapia de reposição onde a proteína FMRP viria de uma fonte externa; possibilidade de reactivar o gene FMR1 e por fim pela psicofarmacologia, que se focaliza no uso de medicamentos mais específicos para atenuar ou eliminar os sintomas da SXF.
8. Síndrome de Apert
|
Descrição Sucinta da Patologia
|
 Patologia que resulta de uma alteração genética, doença autossómica dominante que ocorre no período de fecundação. A causa está numa mutação no cromossoma 10 nos braços q25-q26 do cromossoma, no gene do receptor 2 do factor de crescimento dos fibroblastos (gene FGFR2). O síndrome de Apert compõe as quase seis mil síndromes genéticas conhecidas e é caracterizado por uma má formação do crânio, terço médio da face, mãos e pés e outras diversas alterações funcionais que variam entre indivíduos, onde a maioria dos portadores apresenta deficiência mental com atraso no desenvolvimento.
Patologia que resulta de uma alteração genética, doença autossómica dominante que ocorre no período de fecundação. A causa está numa mutação no cromossoma 10 nos braços q25-q26 do cromossoma, no gene do receptor 2 do factor de crescimento dos fibroblastos (gene FGFR2). O síndrome de Apert compõe as quase seis mil síndromes genéticas conhecidas e é caracterizado por uma má formação do crânio, terço médio da face, mãos e pés e outras diversas alterações funcionais que variam entre indivíduos, onde a maioria dos portadores apresenta deficiência mental com atraso no desenvolvimento.|
Características Gerais (se houver)
|
Os indivíduos com síndrome de Apert apresentam uma anomalia craniofacial chamada de Acrocefalosindactilia Tipo I, consiste numa anomalia craniofacial ou craniossinostose e anomalias das mãos e pés (união dos dedos), que se designa por Sindactilia.
|
Características Orais (se houver)
|
Hiperplasia gengival, apinhamento dentário severo, mordida cruzada, classe III de Angle, postura labial inadequada, fenda palatina, úvula bífida em 30% dos casos, palato alto, macroglossia, erupção tardia e hipoplasia maxilar.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
No que diz respeito ao tratamento, não existe cura, mas é importante a estimulação do desenvolvimento psicomotor e a diminuição das anomalias existentes. Para promover um maior grau de autonomia, a educação física adaptada tem vindo a ser valorizada, e através de tratamentos cirúrgicos pode-se obter um maior grau de funcionalidade e uma aparência menos característica do síndrome. Pode-se efectuar tratamentos cirúrgicos na correcção da sindactilia, correcções dentárias, da fissura palatina e correcção da craniossinostose, antes do 1.º ano de vida, impedindo que as distintas áreas cerebrais sejam afectadas, descomprimindo o espaço interno do crânio.
9. ANEURISMA cerebral
|
Descrição Sucinta da Patologia
|
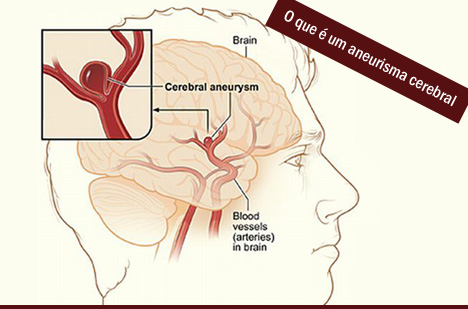 Dilatação do vaso sanguíneo causada pela fragilidade da parede do mesmo. A gravidade do aneurisma depende do seu tamanho e da sua localização. Qualquer vaso pode apresentar uma dilatação aneurismática, porém os dois locais mais comuns são a aorta e os vasos cerebrais.
Dilatação do vaso sanguíneo causada pela fragilidade da parede do mesmo. A gravidade do aneurisma depende do seu tamanho e da sua localização. Qualquer vaso pode apresentar uma dilatação aneurismática, porém os dois locais mais comuns são a aorta e os vasos cerebrais.|
Características Gerais (se houver)
|
Os indivíduos portadores de um aneurisma cerebral só apresentarão características gerais consoante as complicações que advêm da ruptura de um aneurisma, visto que os aneurismas normalmente são assintomáticos.
|
Características Orais (se houver)
|
A patologia não apresenta alterações orais, no entanto estas podem surgir devido à medicação (para controlar as convulsões).
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Quando socorridas atempadamente as pessoas podem minimizar as sequelas. Sendo assim, os tratamentos utilizados são a cirurgia (abertura do crânio e bloqueio do aneurisma com clipes metálicos) e a embolização/tratamento endovascular (inserção de um cateter que vai até ao aneurisma e promove o seu bloqueio com inserção de micro molas de platina).
10. Síndrome de Rett
|
Descrição Sucinta da Patologia
|
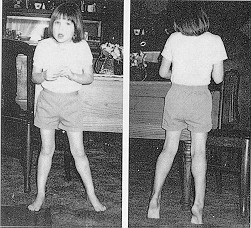 É uma anomalia genética que causa desordens de ordem neurológica quase exclusivamente em crianças do sexo feminino, em todos os grupos étnicos. Compromete progressivamente as funções motora e intelectual originando distúrbios comportamentais e dependência. Após um período de desenvolvimento aparentemente normal – 6 a 18 m. surgem acções estereotipadas (movimentos repetitivos e involuntários das mãos), característica marcante da doença. Está dividido em quatro fases. Mutações no gene que codifica a proteína MeCP2 (proteína responsável pela regulação de vários genes na célula – DNA fica inacessível às proteínas de transcrição e os genes são silenciados). A mutação deste gene origina a expressão excessiva de alguns genes que habitualmente se encontram silenciados. As mutações no gene MECP2 representam uma proporção importante dos casos, mas existirão outros factores ainda não identificados na etiologia desta doença.
É uma anomalia genética que causa desordens de ordem neurológica quase exclusivamente em crianças do sexo feminino, em todos os grupos étnicos. Compromete progressivamente as funções motora e intelectual originando distúrbios comportamentais e dependência. Após um período de desenvolvimento aparentemente normal – 6 a 18 m. surgem acções estereotipadas (movimentos repetitivos e involuntários das mãos), característica marcante da doença. Está dividido em quatro fases. Mutações no gene que codifica a proteína MeCP2 (proteína responsável pela regulação de vários genes na célula – DNA fica inacessível às proteínas de transcrição e os genes são silenciados). A mutação deste gene origina a expressão excessiva de alguns genes que habitualmente se encontram silenciados. As mutações no gene MECP2 representam uma proporção importante dos casos, mas existirão outros factores ainda não identificados na etiologia desta doença.|
Características Gerais (se houver)
|
Os indivíduos portadores do síndrome de Rett nascem e desenvolvem-se normalmente até 6 – 18 meses. Posteriormente deixa de brincar; perde do uso intencional das mãos; tem movimentos manuais atípicos – bater palmas, entrelaçar dedos e mãos, torcer mãos, lavar mãos, mãos na boca; sintomas autisticos; marcha com base alargada; escoliose da coluna vertebral; perda progressiva da comunicação; baixa estatura e crescimento lento da cabeça.
|
Características Orais (se houver)
|
A patologia não apresenta alterações orais, no entanto estas podem surgir devido à medicação (para controlar as convulsões). PB, cálculo, gengivites, cáries e outro tipo de problemas: dependência, falta de destreza; cooperante ou não; tipo alimentação (mole); dificuldades económicas; tipo de tratamento/acompanhamento médico-dentário; frequência da consulta de higiene oral, bruxismo (ranger de dentes).
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Medicação – controlo da mobilidade e controlo das convulsões. Terapia física o mais precoce e intensa possível – prevenir: escolioses; rigidez; pé equino; favorecer a mobilidade. Musicoterapia, aparelhos ortopédicos, massagem subaquática, hidroterapia, hipoterapia. Dar atenção à higiene oral, é importante. Favorecer a marcha, todo o movimento voluntário e insistir sobre a importância de exercícios de reeducação funcional.
11. CRÂNIO BRAQUIOCEFÁLICO/DISPLASIA ÓSSEA E PÉ BOTO BILATERAL
|
Descrição Sucinta da Patologia
|
 A displasia fibrosa é definida como processo patológico benigno em que ocorre a substituição gradativa, do tecido ósseo normal por tecido fibroso e trabéculas ósseas neo-formadas. A sua forma monostótica é a mais comum nos ossos gnáticos, porém quando envolve a maxila pode disseminar-se através dos ossos adjacentes como o zigomático, o esfenóide e o occipital. A displasia fibrosa na região de cabeça e pescoço afecta funcionalmente e esteticamente o paciente portador.
A displasia fibrosa é definida como processo patológico benigno em que ocorre a substituição gradativa, do tecido ósseo normal por tecido fibroso e trabéculas ósseas neo-formadas. A sua forma monostótica é a mais comum nos ossos gnáticos, porém quando envolve a maxila pode disseminar-se através dos ossos adjacentes como o zigomático, o esfenóide e o occipital. A displasia fibrosa na região de cabeça e pescoço afecta funcionalmente e esteticamente o paciente portador. O pé boto caracteriza-se por uma deformidade congénita tridimensional, que afecta 1 a 8 recém-nascidos em cada 1000. Pé boto ou pé zambo é cientificamente denominado de pé equinovaro congénito. O pé adopta uma posição de equinismo, varismo, supinação do retro pé e adução de ante pé. A causa primária do pé equinovaro congénito não está totalmente esclarecida. Aponta-se como causa mais provável uma má posição fetal intra-uterina.
O pé boto caracteriza-se por uma deformidade congénita tridimensional, que afecta 1 a 8 recém-nascidos em cada 1000. Pé boto ou pé zambo é cientificamente denominado de pé equinovaro congénito. O pé adopta uma posição de equinismo, varismo, supinação do retro pé e adução de ante pé. A causa primária do pé equinovaro congénito não está totalmente esclarecida. Aponta-se como causa mais provável uma má posição fetal intra-uterina.|
Características Gerais (se houver)
|
Os indivíduos portadores desta doença apresentam uma discreta elevação dos olhos, descrita como “olhando para o céu” ou “olhar de querubim”. Este aspecto ocular e a face de querubim adquiriram a denominação de querubismo.
O diagnóstico clínico ocorre entre 2 e 4 meses e caracteriza-se por sinais de raquitismo, com achatamento posterior do crânio, espessamento das junções condrocostais, alargamento das porções distais do rádio e do cúbito, aumento da fontanela anterior e das bossas frontais. As fracturas de costelas e ossos longos incidem em 10% dos prematuros de baixo peso. Acompanhando o quadro clínico, pode haver redução no crescimento. Este quadro caracteriza-se como raquitismo da prematuridade, entretanto, o quadro mais frequente é o de hipomineralização, que pode ser evidenciado ao raio X de ossos longos. Nesta fase, quando aparecem sinais radiológicos, já ocorreu redução da densidade óssea com perda de 40% dos minerais do osso.
|
Características Orais (se houver)
|
A patologia não apresenta alterações orais, no entanto estas podem surgir devido à medicação.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Terapia física o mais precoce e intensa possível – prevenir: escolioses; rigidez; pé equino; favorecer a mobilidade. Musicoterapia, aparelhos ortopédicos.
12. SÍNDROME DE WILLIAMS
|
Descrição Sucinta da Patologia
|
 O Síndrome de Williams é uma doença genética de transmissão autossómica dominante. O síndrome deve-se a uma deleção de um gene no cromossoma 7 (7q11.23). Afecta igualmente os dois sexos e causa problemas médicos e do desenvolvimento.
O Síndrome de Williams é uma doença genética de transmissão autossómica dominante. O síndrome deve-se a uma deleção de um gene no cromossoma 7 (7q11.23). Afecta igualmente os dois sexos e causa problemas médicos e do desenvolvimento.|
Características Gerais (se houver)
|
Os indivíduos portadores do Síndrome de Williams apresentam uma face peculiar de “duende”: testa larga, bochechas salientes, nariz com base achatada e extremidade larga, narinas antevertidas, espaço entre o nariz e o lábio superior longo, boca grande, lábios grossos e queixo pequeno; doença cardiovascular associada, défice cognitivo, atraso do desenvolvimento, hiperacúsia, hipercalcémia, artropatia, problemas de coordenação e equilíbrio e dificuldade na alimentação.
|
Características Orais (se houver)
|
Maloclusão, hipoplasia do esmalte, cárie, microdontia, agenesia, exfoliação tardia, diastemas e lábios proeminentes com a boca aberta.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Não existe cura para este síndrome, apenas tratamento mediante a sintomatologia para melhorar a qualidade de vida destes indivíduos. Normalmente a intervenção de vários especialistas como o Cardiologista, Fisioterapeutas, Terapeutas ocupacionais, Médicos dentistas entre outros são importantes.
13. ESCLEROSE TUBEROSA
|
Descrição Sucinta da Patologia
|
Doença genética rara, multi-sistémica que causa tumores benignos no cérebro, rins, coração, olhos, pulmões e pele. Tem igual prevalência em raça, etnia e sexo. Em cada 100.000 nascimentos, 10 a 16 recém-nascidos são afectados.
|
Características Gerais (se houver)
|
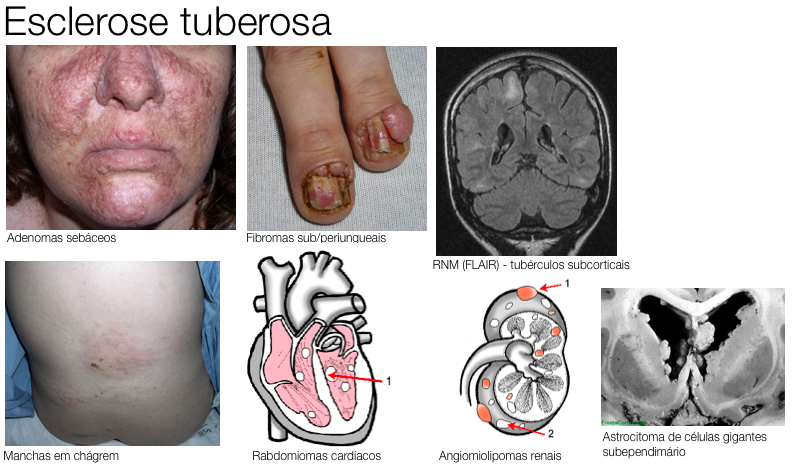 Os sintomas revelam-se, geralmente, no 1.º ano de vida, sendo os mais frequentes: nódulos tuberosos, nódulos subependimários, tumores de células gigantes, angiomiolipomas, rabdomiomas, angiofibromas faciais, máculas hipomelanocíticas, fibromas ungueais e subungueais, placas cutâneas, hamartomas ascrtocíticos, entre outros. 50% dos indivíduos portadores apresentam ainda, dificuldades de aprendizagem moderada a profunda e 80 a 90% epilepsia.
Os sintomas revelam-se, geralmente, no 1.º ano de vida, sendo os mais frequentes: nódulos tuberosos, nódulos subependimários, tumores de células gigantes, angiomiolipomas, rabdomiomas, angiofibromas faciais, máculas hipomelanocíticas, fibromas ungueais e subungueais, placas cutâneas, hamartomas ascrtocíticos, entre outros. 50% dos indivíduos portadores apresentam ainda, dificuldades de aprendizagem moderada a profunda e 80 a 90% epilepsia. |
Características Orais (se houver)
|
Apenas dos medicamentos, normalmente devido a epilepsia associada.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Não tem cura, no entanto existem um conjunto de soluções para o tratamento de sintomas, tais como: cirurgia (angiomiolipomas maiores que 4cm, alguns tumores cerebrais e lesões dermatológicas), homeopatia, fisioterapia, terapia ocupacional e o uso de rapamicina (ainda em estudo).
14. SÍNDROME DA DELECÇÃO DO CROMOSSOMA 22
|
Descrição Sucinta da Patologia
|
Causas: A sua etiologia é desconhecida, sendo esta delecção esporádica em aproximadamente 90% dos casos e herdada de um dos pais em 10% deles.
|
Características Gerais (se houver)
|
Uma das características mais frequentes deste síndrome é a presença de malformação cardíaca, porém outras alterações podem estar presentes, tais como a fenda palatina, hipoplasia do timo e de glândula paratireóide, dismorfias faciais, voz nasalada, dificuldade de aprendizagem, doenças psiquiátricas e atraso mental. O dano causado pelas deleções depende da quantidade e qualidade do material genético perdido, mas no geral, as deficiências são grandes e provocam consequências graves.
|
Características Orais (se houver)
|
Estes indivíduos podem manifestar algumas alterações orais, como maloclusões, anomalias dentárias, falha dentária congénita, dentes pequenos e hipoplasia do esmalte. Podem também apresentar excesso de maxilar vertical, dificuldade em fechar os lábios, face hipotónica com movimentos periorais reduzidos e comissura labial rebaixada.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Fisioterapia, terapia da fala, terapia ocupacional, psicologia individual e familiar, psiquiatria, intervenções cirúrgicas e intervenção medicamentosa.
15. SÍNDROME DE ANGELMAN
|
Descrição Sucinta da Patologia
|
|
Características Gerais (se houver)
|
Atraso na aquisição motora (sentar, gatinhar, andar, etc.); deficiência mental grave; comprometimento ou ausência da fala (98%); andar desequilibrado, com pernas afastadas e esticadas (marcha atáxica); hipotonia; perturbações do sono; hiperactividade; risos imotivados; microcefalia; braquicefalia; movimentos involuntários do tipo mioclónico; convulsões; epilepsia; padrões no EEG característicos; face triangular; hipopigmentação; hipersensibilidade ao calor e gosto especial por água.
|
Características Orais (se houver)
|
Prognatia; protusão lingual e dentes separados.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Não existem tratamentos específicos, mas os sintomas podem ser atenuados por: fisioterapia; dispositivos adaptativos e estimulação intensa da criança.
Quanto aos medicamentos podem ser utilizados antipsicóticos e para a epilepsia, medicamentos anticonvulsivantes.
16. DISTROFIA MIOTÓNICA
|
Descrição Sucinta da Patologia
|
 A Distrofia Miotónica é caracterizada por um grupo de distúrbios que provoca, uma fraqueza muscular progressiva, levando à perda de tecido muscular. Os portadores de Distrofia Miotónica apresentam dificuldades de relaxamento muscular, mas também outros órgãos ou sistemas podem estar afectados, nomeadamente os olhos, coração, sistema endócrino e o sistema nervoso central. Existem 40 tipos de Distrofia Miotónica, o que as diferenciam é a idade em que se manifestam. Sendo o seu prognóstico mais favorável quando o início dos sintomas ocorre após os 20 anos. É uma doença de causa genética que afecta o cromossoma 19. Os sintomas manifestados são fraqueza muscular progressiva, dificuldades em caminhar, retardo intelectual e hipotonia.
A Distrofia Miotónica é caracterizada por um grupo de distúrbios que provoca, uma fraqueza muscular progressiva, levando à perda de tecido muscular. Os portadores de Distrofia Miotónica apresentam dificuldades de relaxamento muscular, mas também outros órgãos ou sistemas podem estar afectados, nomeadamente os olhos, coração, sistema endócrino e o sistema nervoso central. Existem 40 tipos de Distrofia Miotónica, o que as diferenciam é a idade em que se manifestam. Sendo o seu prognóstico mais favorável quando o início dos sintomas ocorre após os 20 anos. É uma doença de causa genética que afecta o cromossoma 19. Os sintomas manifestados são fraqueza muscular progressiva, dificuldades em caminhar, retardo intelectual e hipotonia. |
Características Gerais (se houver)
|
As características manifestadas podem ser: calvície frontal, face longa e estreita, atrofia no músculo temporal, alterações na fala, cataratas, defeitos cardíacos associados, rigidez muscular e incapacidade de relaxamento após a contracção muscular.
|
Características Orais (se houver)
|
Os principais problemas orais encontrados são: deficiente higiene oral, problemas na ATM, maior incidência de cárie, respiração bucal e deglutição atípica. As dificuldades encontradas na consulta de higiene oral são as seguintes: abertura de boca muito limitada, são necessárias muitas pausas para descanso da ATM, não colocar bite-block, idade mental, dificuldades nos movimentos distais (escovagem e fio dentário), distúrbios de deglutição, não inclinar muito a cadeira do paciente e reflexo de vómito.
É importante avaliar a eficácia da escovagem, os ensinos de acordo com o seu nível de compreensão, avaliar a autonomia na sua higiene oral, aplicação de selantes, fluorterapia e orientação na dieta.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
Não há tratamento para a Distrofia Miotónica. O objectivo é controlar os sintomas de modo a aumentar a qualidade de vida. Os portadores de Distrofia Miotónica devem ter acompanhamento de fisioterapeutas.
17. HIDROCEFALIA
|
Descrição Sucinta da Patologia
|
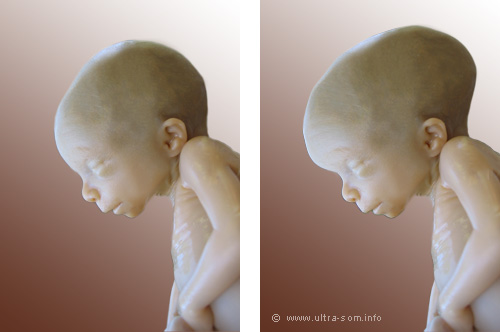 A hidrocefalia é uma acumulação anormal do Liquido Cefalorraquidiano (LCR) nos ventrículos que vai provocar um aumento da pressão intracraniana, consequente aumento de pressão sobre estruturas do cérebro, geralmente causando danos irreversíveis no cérebro como atraso neuro-psico-motor no caso das crianças, que ocorre numa proporção de 1/1000 nascimentos. A hidrocefalia tem três principais causas, aumento da produção de LCR comunicante (extraventricular) onde não existe interferência no fluxo do LCR mas este não é absorvido do espaço subaracnóide que geralmente é a condição mais rara, Não-comunicante (intraventricular/obstrutiva) e a comunicante (extraventricular) onde não existe interferência no fluxo do LCR mas este não é absorvido do espaço subaracnóide. A hidrocefalia congénita caracteriza-se por se manifestar no período peri-natal ou ate ao 1º ano de vida e esta associada a múltiplas causas como malformações congétinas (Malformação de Chiari, estenose do aqueduto de Sylvius), defeitos do tubo neural (Meningomielocele, Espinha Bífida), associado a várias síndromes (Síndrome de Apert, Síndrome de Dandy-Walker, Síndrome de Alexander, entre outros). A adquirida, por traumas, infecções (meningite), hemorragias, prematuridade, anóxia no momento do parto. Pode ainda ser causada por infecções contraídas pela mãe durante a gestação como por exemplo sífilis, rubéola, toxoplasmose, Hepatite B.
A hidrocefalia é uma acumulação anormal do Liquido Cefalorraquidiano (LCR) nos ventrículos que vai provocar um aumento da pressão intracraniana, consequente aumento de pressão sobre estruturas do cérebro, geralmente causando danos irreversíveis no cérebro como atraso neuro-psico-motor no caso das crianças, que ocorre numa proporção de 1/1000 nascimentos. A hidrocefalia tem três principais causas, aumento da produção de LCR comunicante (extraventricular) onde não existe interferência no fluxo do LCR mas este não é absorvido do espaço subaracnóide que geralmente é a condição mais rara, Não-comunicante (intraventricular/obstrutiva) e a comunicante (extraventricular) onde não existe interferência no fluxo do LCR mas este não é absorvido do espaço subaracnóide. A hidrocefalia congénita caracteriza-se por se manifestar no período peri-natal ou ate ao 1º ano de vida e esta associada a múltiplas causas como malformações congétinas (Malformação de Chiari, estenose do aqueduto de Sylvius), defeitos do tubo neural (Meningomielocele, Espinha Bífida), associado a várias síndromes (Síndrome de Apert, Síndrome de Dandy-Walker, Síndrome de Alexander, entre outros). A adquirida, por traumas, infecções (meningite), hemorragias, prematuridade, anóxia no momento do parto. Pode ainda ser causada por infecções contraídas pela mãe durante a gestação como por exemplo sífilis, rubéola, toxoplasmose, Hepatite B.|
Características Gerais (se houver)
|
Os sinais e sintomas da hidrocefalia vão depender da idade em que se manifesta. Em crianças em que as suturas do crânio ainda não estão fechadas nota-se um grande aumento do perímetro cefálico devido à acumulação do LCR, aumento da pressão intracraniana, irritabilidade, atraso neuro-psico-motor, vómitos, fontanela tensa, disjunção das suturas, couro cabeludo adelgaçado com as veias proeminentes.
|
Características Orais (se houver)
|
Não tem características específicas.
|
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
|
O tratamento passa pela redução da acumulação do LCR de modo a manter uma pressão intracraniana estável, evitando assim sequelas neurológicas. Este tratamento baseia-se numa intervenção cirúrgica onde há a implantação de um sistema de drenagem do LCR.
18. SÍNDROME CRI-DU-CHAT
Descrição Sucinta da Patologia
A Síndrome de Cri-du-Chat (CDC) é uma anormalidade cromossómica resultante da deleção de uma porção do braço curto de um dos pares do cromossoma 5. A região que contém as características principais da síndrome está localizada na faixa 5p15.2;A região responsável pelo choro característico da síndrome, está na faixa 5p15.3.
Características Gerais (se houver)
Bebés:
· Choro de “gato”, alto e longo, (devido a má formação da laringe);
· Baixo peso ao nascer;
 · Microcefalia, (cabeça pequena);
· Microcefalia, (cabeça pequena);· “Moonface”;
· Hipertelorismo ocular, (afastamento excessivo dos olhos);
· Pregas epicantais;
· Baixa ponte nasal;
· Prega palmar única (simiesca);
· Desenvolvimento atrasado.
C Crianças e Adultos:
Baixa estatura e magros;
Hipotonia;
Sindactilia nas mãos e nos pés,(união entre 2 ou mais dedos);
Baixa implantação das orelhas;
Aracnodactilia, (dedos longos);
Atraso mental.
Hipertelorismo ocular;
Pregas epicantais;
Características Orais (se houver)
Dificuldade a sugar e a engolir;
Apresentam refluxo gástrico;
Retrognatismo;
Dentes projectados para a frente;
Micrognastia, (mandíbula pequena).
Tratamentos (medicação, cirurgias, fisioterapia, etc. – para redução dos sintomas)
Não tem Cura;
O tratamento só pode ajudar as crianças a desenvolverem-se o melhor possível;
Importante intervenção de:
÷ Terapeuta da Fala;
÷ Fisioterapeutas;
÷ Terapeutas ocupacionais;
÷ Higienistas Orais.
Para algumas crianças, as drogas têm sido de grande auxílio para problemas relacionados com o sono e para controlar os seus comportamentos.
O quanto o indivíduo é afectado não está directamente relacionado com a quantidade de material genético perdido; Incentivar o potencial de desenvolvimento da criança ao máximo; Fazer o máximo para que leve a vida mais normal possível.
19. Síndrome de Sotos
|
Descrição sucinta da patologia
|
 O Síndroma de Sotos também conhecido como Gigantismo Cerebral é uma patologia de etiologia genética (5q35), mais comum no sexo masculino com implicações ao nível do:
O Síndroma de Sotos também conhecido como Gigantismo Cerebral é uma patologia de etiologia genética (5q35), mais comum no sexo masculino com implicações ao nível do:Complexo Crânio-Facial:macrocrânio com dolicocefalia (cabeça alongada no sentido vertical), hipertelorismo ocular (olhos demasiado afastados), problemas oculares, auditivos, linha de implantação do cabelo recuada, testa proeminente, palato alto e estreito, prognatismo mandibular.
Sistema Nervoso: disfunção neurológica não-progressiva, ventrículos cerebrais dilatados, distúrbios comportamentais, ataraso no desenvolvimento psico-motor, convulsões e hipotonia.
Sistema Esquelético: bebés grandes ao nascimento (perímetro cefálico normalmente aumentado, idade óssea 2 a 3 anos mais avançada que a idade cronológica, mãos e pés grandes, escoliose e pé chato.
|
Características gerais (se houver)
|
- Crescimento precoce excessivo
- Maturação Óssea precoce
- Hipotonia
- Características faciais particulares
|
Características orais (Não há) Ter Em Atenção Na Consulta Ho
|
Características Orais: Palato alto e estreito, Erupção dentária precoce, Prognatismo mandibular.
Ter em atenção na consulta: distúrbios comportamentais (ansiedade, fobias, irritabilidade) Hipotonia (problemas com a alimentação, dificuldades respiratórias…)
Patologias Médicas que podem estar associadas: cardiopatias congénitas, patologias renais, epilepsia, infecções recorrentes, atraso no desenvolvimento psico-motor.
|
Tratamentos (medicação cirurgias fisio etc para redução dos sintomas)
|
Não há tratamento. Nos primeiros anos de vida a intervenção médica é feita principalmente para diminuir os problemas com a alimentação. Tenta-se prevenir precocemente os problemas comportamentais e monitorizar o crescimento.
20. SINDROME DE KLINEFELTER
|
Descrição sucinta da patologia
|
 A síndrome de Klinefelter é: restrita aos homens, causada por uma aneuploidia dos cromossomas sexuais (alteração cromossômica numérica) na maioria dos casos, a um cariótipo 47,XXY.
A síndrome de Klinefelter é: restrita aos homens, causada por uma aneuploidia dos cromossomas sexuais (alteração cromossômica numérica) na maioria dos casos, a um cariótipo 47,XXY. Forma de hipogonadismo masculino caracterizada pela presença de um cromossoma. Como têm um cromossomo Y, estes indivíduos apresentam fenótipo masculino,
As anormalidades variam com o número de cromossomas X adicionais.
|
Características gerais (se houver)
|
Esterilidade
Desenvolvimento de seios (Ginecomastia)
Características masculinas incompletas
Problemas sociais e/ou de aprendizagem
|
Características orais (Não há) Ter Em Atenção Na Consulta Ho
|
Acidentes vasculares cerebrais: Tomam anticoagulantes, possivel cobertura antibiotica (prevenir pneumonia por aspiração), paralisia orofacial unilateral, dificuldade no controlo das secreções, disfagia, cáries, periodontopatias, halitose e disfagia. Cancro da mama: Quimio e radioterapia levam à diminuição de células sanguineas; ↑ do risco de ; hemorragias, anemias, infecções e fadiga, mucosites, xerostomia disgeusia, cáries, d. period, nauseas, vómitos. Atraso motor: Falta de distreza manual. Défice auditivo,: Não compreenção dos ensinos. Doenças auto-imunes(diabetes mellitus; doenças do colagéneo): Saber se tomou a medicação, se comeu antes da consulta, valores de glicémia..
|
Tratamentos (medicação cirurgias fisio etc para redução dos sintomas)
|
Não há tratamento. Deve ser medido periodicamente o nível sérico dos hormônios sexuais. Para o metabolismo com baixo nível de hormônios, o tratamento com hormônios sexuais masculinos pode ser benéfico.O mais comum é a administração uma vez por mês de Depotestosterona injetável, (forma sintética de testosterona). Com a idade a dose e frequencia são aumentadas gradualmente progressão normal do desenvolvimento físico e sexual, incluindo:
crescimento de pêlos pubianos, aumento do tamanho do pênis e testículos, crescimento da barba, desenvolvimento da voz grave,aumento da força muscular.

Muito bom!
Dá sempre uma importante ajuda na clinica a 2ºf.
Abraço Celso
JPG
GostarGostar